Client
Triveni Bio: “We are pioneering a genetics-informed precision medicine approach to develop functional antibodies for the treatment of immunological and inflammatory (I&I) disorders. Using our proprietary approach, we hunt for, and derisk, novel therapeutic targets with strong genetic evidence across diseases. Furthermore, we predict and stratify patient subpopulations, and we provide insights into disease mechanisms.”
Background & Challenge
In early drug discovery, choosing the right target can make or break a development project - even if failure only becomes apparent years later. Genetic evidence plays a key role in this decision: drug targets with genetic support are three times less likely to be abandoned in later development stages [1], making the accurate interpretation of genetic data a priority for any target identification workflow.
Genome-wide association studies (GWAS) are a powerful resource for identifying genomic variants associated with diseases and traits. However, most genetic variants fall outside protein-coding regions, meaning GWAS results alone rarely point directly to a causal gene. Instead, the genomic region surrounding a variant may contain several, or even many, candidate genes, each of which could plausibly be affected. Gene prioritization is the process of ranking these candidates to identify the most likely causal gene driving the association signal. Since this step links a target gene to a genetic variant and therefore a specific disease, it is a highly important step in the target selection process.
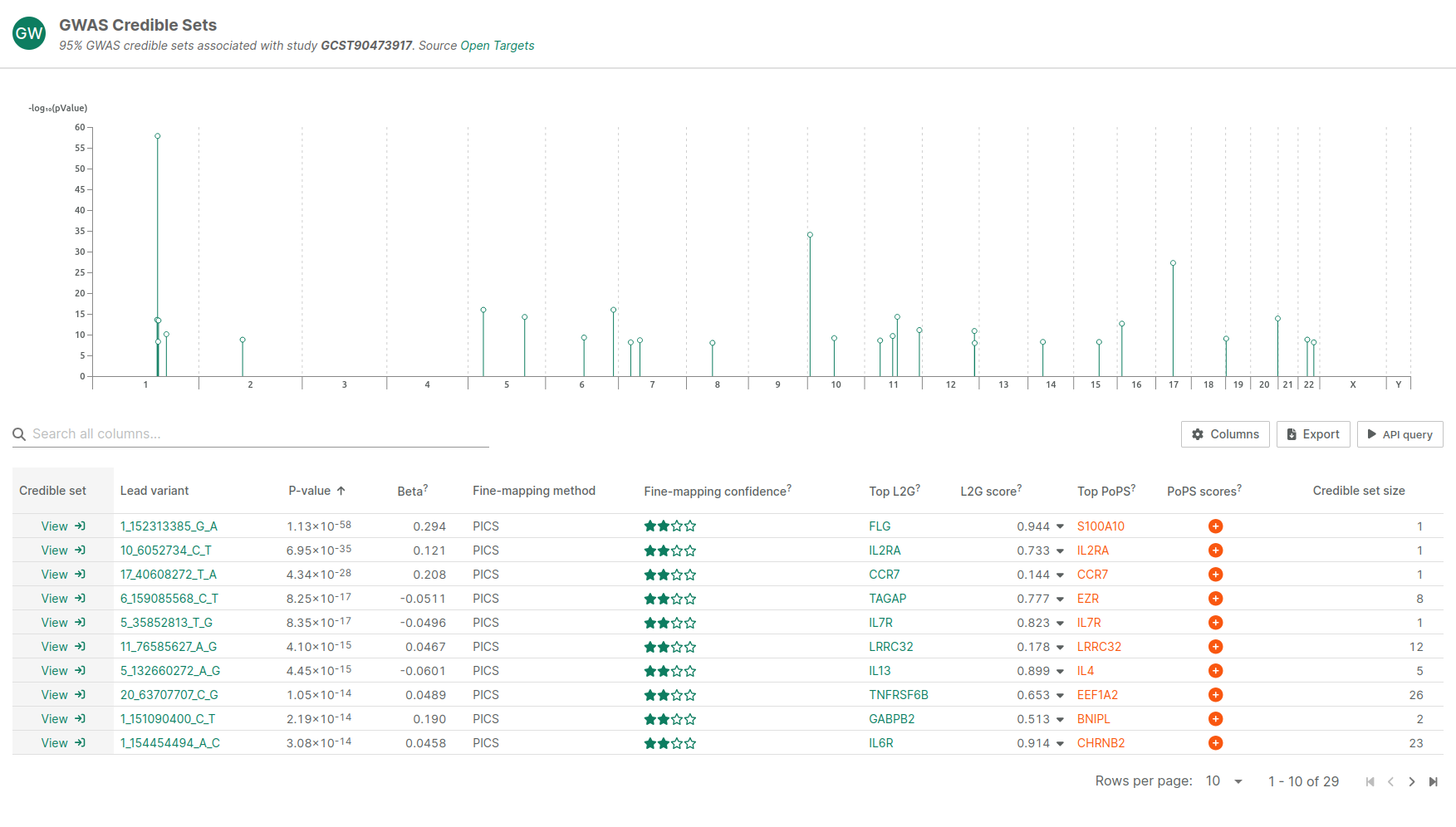
Triveni Bio tasked The Hyve with enhancing their gene prioritization workflow by running a recently developed method called Polygenic Priority Score (PoPS) [2] on all GWAS studies in the Open Targets Genetics portal [3]. To explore the performance improvements by combining PoPS with other gene prioritization methods, including Locus-to-Gene and nearest gene, The Hyve performed comparisons of these methods. Additionally, results of the methods were included in Triveni Bio's internal Open Targets instance (Figure 1), allowing Triveni Bio's researchers to draw directly on multiple complementary sources of genetic evidence.
Our Approach
The Hyve set out to expand Triveni Bio's genetic evidence pipeline, which is based on the Open Targets Genetics portal. To scale PoPS across the 9,000 GWAS studies available in the Open Targets Genetics portal, we integrated PoPS into a workflow management system, enabling automated, reproducible, and scalable execution. We did this in collaboration with engineers from Datapol, who handled the creation of the final pipeline and running it in a scalable manner in Triveni Bio’s cloud environment.
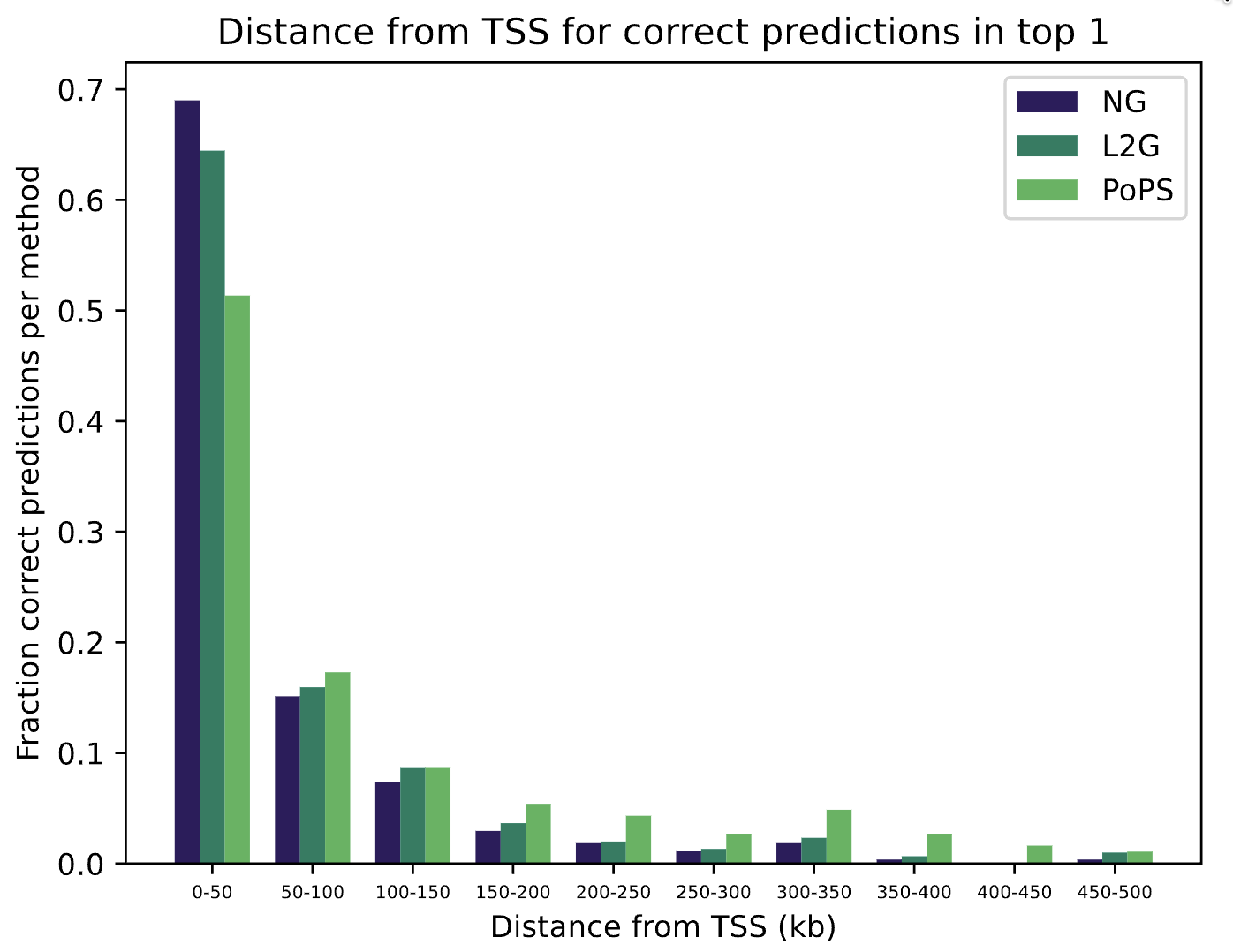
To benchmark PoPS against L2G, the gene prioritization method native to Open Targets that integrates fine-mapping, QTL colocalization, and distance-based metrics [3], we evaluated both methods against two independent gold standard datasets of loci with experimentally validated causal genes. We additionally used a simple distance metric that measures the distance from the variant to the transcription start site of a gene, which we refer to as Nearest Gene. For each locus, candidate genes were ranked by each method, and recall was calculated using a rank-based threshold to determine whether the causal gene was correctly prioritized. This analysis revealed complementary strengths between the methods: L2G showed stronger performance for variants with causal genes in close proximity, consistent with its reliance on QTL colocalisation and positional features, while PoPS more frequently prioritised distal causal genes by leveraging functional genomics signatures and gene network context (Fig. 2). This distinction is particularly relevant in drug discovery, where causal genes are not always the nearest gene to the associated variant, and missing distal candidates may result in overlooking promising therapeutic targets.
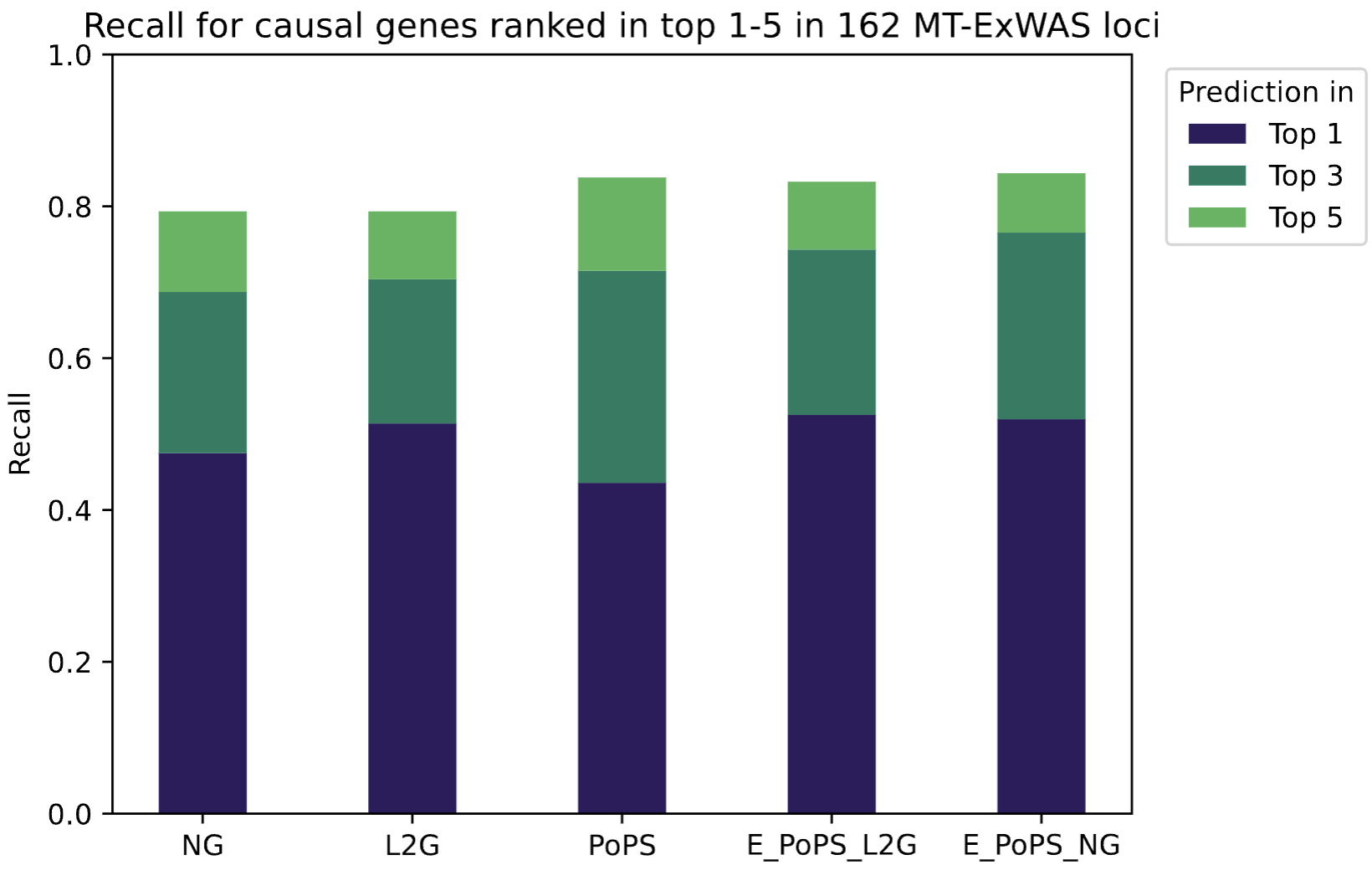
To leverage the complementary strengths of both methods, we trained a simple machine learning model using PoPS and L2G scores as input features. The combined model outperformed either method in isolation, achieving higher recall for causal gene identification across both gold standard datasets (Fig. 3). Interestingly, a model trained with PoPS and Nearest Gene as features performs slightly better. This demonstrates that the two types of methods, functional genomics-based (PoPS), and variant centric-based (L2G and Nearest Gene), capture partially independent signals from the underlying GWAS data, and that their integration yields a more robust prioritization of disease-relevant genes. For drug discovery applications, this improved prioritization directly translates to a higher-confidence set of therapeutic targets for further experimental validation and pipeline progression.
Results & Next Steps
In this project, Triveni Bio and The Hyve demonstrated that integrating complementary gene prioritization methods improves the identification of causal genes from GWAS data. By scaling PoPS across 9,000 studies in the Open Targets Genetics portal and combining it with the Nearest Gene method through an ensemble model, Triveni Bio now has access to a more comprehensive and higher-confidence gene prioritization workflow. The performance gains observed across the gold standard benchmarks suggest that this approach can reduce the risk of overlooking true causal genes - a critical consideration in early-stage target selection for immunological and inflammatory diseases.
Looking ahead, work is ongoing to integrate PoPS directly into the Open Targets Gentropy pipeline, the recently developed post-GWAS analysis pipeline developed by the Open Targets Consortium. This will enable PoPS scores to be updated systematically as new GWAS data becomes available, further embedding genetic evidence into Triveni Bio's target identification workflow.
Want to see PoPs in action? Visit our demo server to explore how PoPS and L2G scores complement one another (Fig. 1). You can also explore additional capabilities we have built into the Open Targets Platform, including literature summarisation, cBioPortal custom data integration, and knowledge graph representations of the data — all designed to support evidence-based target identification.
References
[1] Minikel, E.V., Painter, J.L., Dong, C.C. et al. Refining the impact of genetic evidence on clinical success. Nature 629, 624–629 (2024). https://doi.org/10.1038/s41586-024-07316-0
[2] Weeks, E.M., Ulirsch, J.C., Cheng, N.Y. et al. Leveraging polygenic enrichments of gene features to predict genes underlying complex traits and diseases. Nat Genet 55, 1267–1276 (2023). https://doi.org/10.1038/s41588-023-01443-6
[3] Mountjoy, E., Schmidt, E.M., Carmona, M. et al. An open approach to systematically prioritize causal variants and genes at all published human GWAS trait-associated loci. Nat Genet 53, 1527–1533 (2021). https://doi.org/10.1038/s41588-021-00945-5
[4] Schipper, M., de Leeuw, C.A., Maciel, B.A.P.C. et al. Prioritizing effector genes at trait-associated loci using multimodal evidence. Nat Genet 57, 323–333 (2025). https://doi.org/10.1038/s41588-025-02084-7